Author: V. Dimov, M.D., Allergist/Immunologist at Cleveland Clinic
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
A 72-year-old male was treated for multiple meyeloma 2 years ago. Subsequently, his IgG was found to be 180 mg/dL, IgA 7, IgM and there was insufficient response on the pneumococcus serotypes. At an outside facility, SCIG was started due to poor IV access. There was no history of infections. The diagnosis was secondary immunodeficiency. He has been receiving SCIG (Hizentra) 25 gm every week.
A year later, his IgG is 1200 mg/dL. The reported last dose of SCIG was 3-4 weeks ago (the prescription ran out).
What dose adjustment would you recommend now?
SCIG dose adjustement:
Based on the typical IVIG starting dose of 400 mg/kg/month, the total monthly dose is 400 mg x 65 kg = 26,000 mg (26 g).
According to the Hizentra dosage calculator (Initial recommended dose of Hizentra = 1.53 x Previous IVIg dose(grams)
Number of weeks between scheduled IVIg doses). Based on an IVIg dose of 26grams and 4weeks between IVIg doses, Hizentra weekly dose is: 9.95 g (49.73).
Patient's states the last SCIG dose was 3-4 weeks ago, total IgG is 1200 mg/dL. It is possible that some of the production of IgG by the B cells is recovering.
The recommended new weekly dose of SCIG is 15 g every week. IgG, IgM, IgA levels should be checked in 3 months, just before the SCIG infusion.
SCIG prescription was changed. The infusion nurse was informed.
Mnemonic: Dose of IVIG in PIDD: 4
400-600 mg/kg/month
IgG trough level should be over 400 mg/dL (over 600 mg/dL if bronchiectasis)
4 letter words:
IVIG
CVID
SCID
Starting doses for IVIG: 400 to 600 mg/kg/month for a target trough level of at least 500 mg/dL.
How monitor CVID patients on IVIG?
Immunoglobulin levels at 3-6 month intervals
CBC and CMP yearly
Spirometry yearly
CT chest every 3-4 years - only if lung disease suspected or lung functions not normal
References
Guidelines on Dosing and Treatment Administration for Hizentra Therapy http://buff.ly/1uCZt4Q
Related reading
AInotes - Common Variable Immunodeficiency http://buff.ly/1uCZFky
AInotes - IVIG and Subcutaneous Ig http://buff.ly/1uCZTYP
Published: 07/12/2010
Updated: 03/15/2014
Showing posts with label CVID. Show all posts
Showing posts with label CVID. Show all posts
Follow-up of a Patient with Common Variable Immune Deficiency (CVID)
Author: V. Dimov, M.D., Allergist/Immunologist and Assistant Professor at University of Chicago
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at LSU (Shreveport) Department of Allergy and Immunology
A 34-year-old female is in the Allergy/Immunology clinic today for a follow-up of Common Variable Immune Deficiency (CVID). She was last seen in the clinic 3 months ago. Since then, her condition has remained stable and the level of immunoglobulin G has been higher than 700. She is receiving IVIG 90 gm IV every month. She is pre-medicated with Tylenol and oral antihistamines and is able to tolerate the infusions well and has not had any infections of the sinuses or lung infections recently. She however reports painful area in her left armpit for the past two weeks, and it looks like she is developing hidradenitis.
Past medical history (PMH)
Common Variable Immune Deficiency (CVID).
Physical examination
The physical examination is positive for induration and erythema in the right axia which was diagnosed as hidradenitis. The rest of her examination is normal.
What is the most likely diagnosis?
This is a patient with CVID which has been stable on the IVIG replacement at the dose of 90 gm of Gammaguard every month. She also has hidradenitis in the left axilla.
What would you do?
Continue Gammaguard at a dose of 90 grams IV every month. Repeat the level in three months. For her hidradenitis, we recommended Keflex 500 mg PO B.I.D. x 7days.
The patient is planning a trip to the Bahamas for a week in the coming month. She wants to know if she should take any antibiotics with her.
What antibiotics would you prescribe, if she needs any?
Regarding her trip to the Bahamas: in case she develops respiratory or gastrointestinal infection we provided her with a prescription for Levaquin 500 PO QD x 7 days. She is to return to the clinic in four months.
What are the normal serum immunoglobulin levels (IgG, IgA, IgM)?
Serum levels of IgM, IgG and IgA vary with age, gender and race.
The IgG and IgA concentrations in children show a gradual rise with increasing age. The IgA level is generally about the same in both sexes. Girls typically have higher IgM and IgG levels than boys.
The confidence interval bounded by two standard deviations about the mean excludes 5% of apparently healthy controls.
Elevated IgM, low IgA, low IgG, low IgM, and elevated IgA are the commonest changes observed in apparently healthy humans.
Humoral immunodeficiency is commonly defined as IgG, IgM or IgA level that is two standard deviations (2 SD) below the mean level for IgG, IgM or IgA, respectively, for the particular age group and gender.
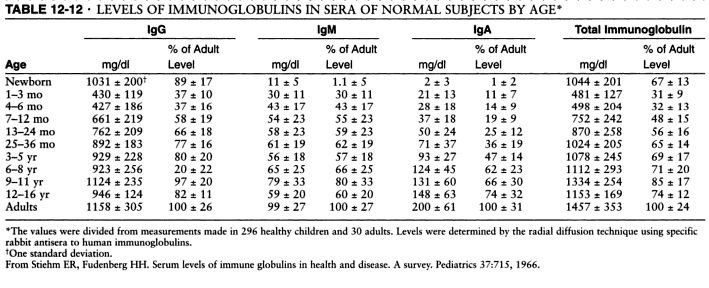
Serum levels of IgM, IgG and IgA. Source: Pediatrics, 1966 and Immunologic disorders in infants and children, by E. Richard Stiehm, Hans D. Ochs, Jerry A. Winkelstein.
Typical pattern of immunoglobulin levels (IgG, IgA, IgM) in humoral immunodeficiency. Click here to enlarge the table.
References
Diagnostic Criteria for Common Variable Immunodeficiency (CVID): Probable and Possible Diagnosis
The diagnostic criteria are divided into three categories: definitive, probable, and possible. There are no criteria for definitive diagnosis of Common Variable Immunodeficiency (CVID) at this time.
To guard against the inclusion of patients who have polymorphic variants in the genes associated with immunodeficiency and to specify the clinical or laboratory finding that is most consistently abnormal in a particular disorder, the patient must fulfill an inclusion criterion that is characteristic of the disorder.
Definitive diagnosis
Patients with a definitive diagnosis are assumed to have a greater than 98% probability that in 20 years they will still be given the same diagnosis. Mutation detection is the most reliable method of making a diagnosis but a single mutation is rarely found in CVID.
Probable diagnosis
Patients with a probable diagnosis are those with all of the clinical and laboratory characteristics of a particular disorder but who do not have a documented abnormality in the gene, the mRNA, or the protein that is known to be abnormal in the disorder. They are assumed to have a greater than 85% probability that in 20 years they will be given the same diagnosis.
Probable diagnosis of CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in serum IgG AND IgA and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Possible diagnosis
Patients with a possible diagnosis are those that have some but not all of the characteristic clinical or laboratory findings of a particular disorder.
Possible diagnosis if CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in one of the major isotypes (IgM, IgG, and IgA) and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Clinical features of CVID
Most patients with CVID are diagnosed with immunodeficiency in the second, third, or fourth decade of life, after they have had several pneumonias; however, children and older adults may be affected.
Viral, fungal, and parasitic infections as well as bacterial infections may be found.
The serum concentration of IgM is normal in about half of the patients.
Abnormalities in T cell numbers or function are common. The majority of patients have normal numbers of B cells; however, some have low or absent B cells.
Approximately 50% of patients have autoimmune manifestations. There is an increased risk of malignancy.
Differential diagnosis of hypogammaglobulinemia includes drug-induced, for example secondary to glucocorticoids (steroids).
Diagnostic Criteria for Primary Immunodeficiencies. Mary Ellen Conley, Luigi D. Notarangelo, and Amos Etzioni Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clinical Immunology, Vol. 93, No. 3, December, pp. 190–197, 1999.
Published: 05/12/2010
Updated: 11/23/2011
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at LSU (Shreveport) Department of Allergy and Immunology
A 34-year-old female is in the Allergy/Immunology clinic today for a follow-up of Common Variable Immune Deficiency (CVID). She was last seen in the clinic 3 months ago. Since then, her condition has remained stable and the level of immunoglobulin G has been higher than 700. She is receiving IVIG 90 gm IV every month. She is pre-medicated with Tylenol and oral antihistamines and is able to tolerate the infusions well and has not had any infections of the sinuses or lung infections recently. She however reports painful area in her left armpit for the past two weeks, and it looks like she is developing hidradenitis.
Past medical history (PMH)
Common Variable Immune Deficiency (CVID).
Physical examination
The physical examination is positive for induration and erythema in the right axia which was diagnosed as hidradenitis. The rest of her examination is normal.
What is the most likely diagnosis?
This is a patient with CVID which has been stable on the IVIG replacement at the dose of 90 gm of Gammaguard every month. She also has hidradenitis in the left axilla.
What would you do?
Continue Gammaguard at a dose of 90 grams IV every month. Repeat the level in three months. For her hidradenitis, we recommended Keflex 500 mg PO B.I.D. x 7days.
The patient is planning a trip to the Bahamas for a week in the coming month. She wants to know if she should take any antibiotics with her.
What antibiotics would you prescribe, if she needs any?
Regarding her trip to the Bahamas: in case she develops respiratory or gastrointestinal infection we provided her with a prescription for Levaquin 500 PO QD x 7 days. She is to return to the clinic in four months.
What are the normal serum immunoglobulin levels (IgG, IgA, IgM)?
Serum levels of IgM, IgG and IgA vary with age, gender and race.
The IgG and IgA concentrations in children show a gradual rise with increasing age. The IgA level is generally about the same in both sexes. Girls typically have higher IgM and IgG levels than boys.
The confidence interval bounded by two standard deviations about the mean excludes 5% of apparently healthy controls.
Elevated IgM, low IgA, low IgG, low IgM, and elevated IgA are the commonest changes observed in apparently healthy humans.
Humoral immunodeficiency is commonly defined as IgG, IgM or IgA level that is two standard deviations (2 SD) below the mean level for IgG, IgM or IgA, respectively, for the particular age group and gender.

Serum levels of IgM, IgG and IgA. Source: Pediatrics, 1966 and Immunologic disorders in infants and children, by E. Richard Stiehm, Hans D. Ochs, Jerry A. Winkelstein.
Typical pattern of immunoglobulin levels (IgG, IgA, IgM) in humoral immunodeficiency. Click here to enlarge the table.
References
Serum immunoglobulin levels in healthy children and adults. J. W. Stoop, B. J. M. Zegers, P. C. Sander, and R. E. Ballieux. Clin Exp Immunol. 1969 January; 4(1): 101–112.
The relationship of race, sex, and age to concentrations of serum immunoglobulins expressed in international units in healthy adults in the USA. S. E. Maddison, C. C. Stewart, C. E. Farshy, and C. B. Reimer. Bull World Health Organ. 1975; 52(2): 179–185.
Serum immunoglobulin concentrations in preschool children measured by laser nephelometry: reference ranges for IgG, IgA, IgM. D Isaacs, D G Altman, C E Tidmarsh, H B Valman, and A D Webster. J Clin Pathol. 1983 October; 36(10): 1193–1196.
D Isaacs, A D Webster, and H B Valman. Clin Exp Immunol. 1984 November; 58(2): 335–340.
Serum Immunoglobulin Levels Throughout the Life-Span of Healthy Man. Ann of Int Med, November 1, 1971, Vol. 75 no. 5 673-682.
Diagnostic Criteria for Common Variable Immunodeficiency (CVID): Probable and Possible Diagnosis
The diagnostic criteria are divided into three categories: definitive, probable, and possible. There are no criteria for definitive diagnosis of Common Variable Immunodeficiency (CVID) at this time.
To guard against the inclusion of patients who have polymorphic variants in the genes associated with immunodeficiency and to specify the clinical or laboratory finding that is most consistently abnormal in a particular disorder, the patient must fulfill an inclusion criterion that is characteristic of the disorder.
Definitive diagnosis
Patients with a definitive diagnosis are assumed to have a greater than 98% probability that in 20 years they will still be given the same diagnosis. Mutation detection is the most reliable method of making a diagnosis but a single mutation is rarely found in CVID.
Probable diagnosis
Patients with a probable diagnosis are those with all of the clinical and laboratory characteristics of a particular disorder but who do not have a documented abnormality in the gene, the mRNA, or the protein that is known to be abnormal in the disorder. They are assumed to have a greater than 85% probability that in 20 years they will be given the same diagnosis.
Probable diagnosis of CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in serum IgG AND IgA and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Possible diagnosis
Patients with a possible diagnosis are those that have some but not all of the characteristic clinical or laboratory findings of a particular disorder.
Possible diagnosis if CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in one of the major isotypes (IgM, IgG, and IgA) and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Clinical features of CVID
Most patients with CVID are diagnosed with immunodeficiency in the second, third, or fourth decade of life, after they have had several pneumonias; however, children and older adults may be affected.
Viral, fungal, and parasitic infections as well as bacterial infections may be found.
The serum concentration of IgM is normal in about half of the patients.
Abnormalities in T cell numbers or function are common. The majority of patients have normal numbers of B cells; however, some have low or absent B cells.
Approximately 50% of patients have autoimmune manifestations. There is an increased risk of malignancy.
Differential diagnosis of hypogammaglobulinemia includes drug-induced, for example secondary to glucocorticoids (steroids).
References
Diagnostic Criteria for Primary Immunodeficiencies. Mary Ellen Conley, Luigi D. Notarangelo, and Amos Etzioni Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clinical Immunology, Vol. 93, No. 3, December, pp. 190–197, 1999.
Recognizing Primary Immune Deficiency in Clinical Practice. Clinical and Vaccine Immunology, March 2006, p. 329-332, Vol. 13, No. 3.
Frequency of follow-up visits in a patient receiving intravenous immunoglobulin (IVIG) infusions - every 6 months? AAAAI Ask the Expert, 2011.
Outcome of allogeneic stem cell transplantation (ASCT) in adults with common variable immunodeficiency (CVID) (JACI, 2011).
Frequency of follow-up visits in a patient receiving intravenous immunoglobulin (IVIG) infusions - every 6 months? AAAAI Ask the Expert, 2011.
Outcome of allogeneic stem cell transplantation (ASCT) in adults with common variable immunodeficiency (CVID) (JACI, 2011).
Published: 05/12/2010
Updated: 11/23/2011
Primary immunodeficiency disorders (PIDD)
Author: V. Dimov, M.D., Allergist/Immunologist and Assistant Professor at University of Chicago
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
Since Bruton described X-linked agammaglobulinemia in 1952, more than 180 different primary immunodeficiencies have been described.
10 Warning Signs of Primary Immunodeficiency in Children
1. Four or more new ear infections within one year.
2. Two or more sinus infections within one year, or chronic sinusitis.
3. Two or more months on antibiotics with little effect.
4. Two or more pneumonias within one year, or pneumonia twice over any time.
5. Failure of an infant to gain weight or grow normally.
6. Recurrent, deep skin or organ abscesses.
7. Persistent thrush in mouth or fungal infection on skin.
8. Need for intravenous antibiotics to clear infection.
9. Two or more deep-seated infections including septicemia.
10. A family history of primary immunodeficiency.
10 Warning Signs of Primary Immunodeficiency in Adults
1. Two or more new ear infections within one year.
2. Two or more serious sinus infections within one year, in the absence of an allergy; or chronic sinusitis.
3. One pneumonia per year for more than one year, or pneumonia twice over any time.
4. Chronic diarrhea with weight loss.
5. Recurrent viral infections (colds, herpes, warts, condyloma).
6. Recurrent need for intravenous antibiotics to clear infections.
7. Recurrent, deep abscesses of the skin or internal organs.
8. Persistent thrush or fungal infection on skin or elsewhere.
9. Infection with normally harmless tuberculosis-like bacteria.
10. A family history of primary immunodeficiency.
Source: JMFworld.com. PDF handouts: Children PIDD, Children PIDD (illustrated), Adult PIDD, 4 Stages of Immunologic Testing when PIDD is suspected.

Primary immunodeficiency disorders (PIDD) (click to enlarge the image).
Definition of PIDD: Genetically determined immunodeficiency.
The World Health Organization recognizes more than 100 PIDD. Most of PIDD are rare but selective IgA deficiency is relatively common, prevalence 1:500.
4 Major Host Defense Mechanisms:
- B-cell immunity
- T-cell immunity
- Phagocytic cells
- Complement system
Phagocyte immunodeficiencies (click to enlarge the image):

Read more:
Phagocyte Deficiencies
Chronic Granulomatous Disease (CGD)
Chediak-Higashi Syndrome (CHS)
Leukocyte adhesion deficiency (LAD)
Leukocyte adhesion deficiency type I (LAD I)
Leukocyte adhesion deficiency type II (LAD II)
Leukocyte adhesion deficiency type III (LAD III)
Hyper IgE Syndrome (HIES)

Humoral immunodeficiency (click to enlarge the image).
B and T cells "talk" constantly in T-dependent immune responses where a host of enzymes are involved in class switching - AICD, UNG, CD40, CD40L - if there is a defect in any of these enzymes, B cells cannot class switch - Ig gets "stuck" at IgM level and hyper-IgM immonodeficiency develops.

T-cell Immunodeficiencies (click to enlarge the image).
The autoimmune regulatory gene (AIRE) is expressed in the thymus. AIRE promotes expression of non-thymic tissue antigens in the thymus - a key part of the thymic education of T cells.
Mutation in the AIRE gene produces disorders such as autoimmune polyglandular syndrome or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). In this syndrome, T cells do not develop tolerance to self-antigens. The endocrine organs are attacked by autoreactive T cells. The AIRE mutation was the first report of a single-gene defect causing a systemic human autoimmune disease.
Mutations in FAS or caspase 10 manifest as autoimmune lymphoproliferative syndrome (ALPS). In ALPS, lymphocytes do not get a signal to "die" and they accumulate in the lymphoid organs.
Read more:
IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X linked) syndrome
Autoimmune lymphoproliferative syndrome (ALPS)
Chronic Mucocutaneous Candidiasis (CMCC)
Diagnosis of T-cell Immunodeficiency

Combined Immunodeficiencies (click to enlarge the image).
Bare lymphocyte syndromes include MHC class I and MHC class II deficiencies. These are primary immune deficiency disorders (PIDD) due to a lack of expression of either MHC I or MHC II. MHC class I deficiency leads to CD8 lymphopenia. MHC class II deficiency leads to CD4 lymphopenia.
Read more:
DiGeorge Syndrome (DGS)
Wiskott-Aldrich Syndrome (WAS)
Ataxia-Telangiectasia (A-T)
.jpg)
Severe Combined Immunodeficiency (SCID) (click to enlarge the image).
In SCID, the younger the age of the patient at the time of transplantation, the better the prognosis. There is a 95% survival rate in an infant who undergoes a transplant before 3 months of age. After six months, the survival rate decreases dramatically, to 50%.
Receptor editing reactivates RAG-1 and RAG-2 when a high affinity self-antigen is recognized by a B cell receptor (BCR). RAG-1 and RAG-2 defects lead to Omenn syndrome, a form of SCID.

Severe combined immunodeficiency (SCID) - 4 groups according to T/B/NK cells (click to enlarge the image).
Receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 contain γ chain, which is affected in X-linked SCID.

Cytokine receptors (click to enlarge the image)

Complement deficiencies (click to enlarge the image).
Read more in Complement Deficiencies.
Tests and Workup of Suspected Immunodeficiency - mostly primary immunodeficiency disorders (PIDD)
B-cell PIDD
Flow Cytometry: CD19 and CD20
Ig G, A, M, E
IgG subclasses (IgG1, IgG2, IgG3, IgG4)
IgG titer for Tetanus
IgG titer for Diphtheria
IgG titer for Haemophilus influenzae type B
IgG titer for Mumps
IgG titers for Pneumococcus
IgG titers for Pneumococcal conjugated vaccine (if given PCV7 or PCV13, Prevnar in the U.S.)
IgG titers for Pneumococcal unconjugated vaccine (if given PSPV, Pneumovax in the U.S.)
Blood group Isohemagglutinins (for diagnosis of CVID)
Mitogen stimulation assays for B- and T-cells
T-cell PIDD (T-cell and NK cell)
Absolute lymphocyte count (CBC-Diff)
Flow Cytometry: CD3CD4, CD3CD8, and CD16CD56
Delayed-type hypersensitivity
Enzyme assays (ADA and PNP)
NK cytolysis assay
Mitogen stimulation assays for B- and T-cells
Phagocyte PIDD (neutrophils, macrophages and monocytes)
CBC-Diff with peripheral smear
Absolute neutrophil count
Flow Cytometry: LFA-1 (CD11a or CD18) and CD15
Oxidative function (DHR or NBT or chemiluminescence)
Enzyme assays (MPO and G6PDH)
Phagocyte function assay - chemotaxis and bactericidal function
Complement PIDD
C3 and C4
C1q
C1 esterase inhibitor (C1-INH) - qualitative and quantitative
CH50 (classic pathway)
AH50 (alternative pathway)
References
Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 2005 May;94(5 Suppl 1):S1-63.
Algorithm 1: General Approach for the Diagnosis of Primary Immunodeficiency
Algorithm 2: Diagnosis of Humoral Immunodeficiency
Algorithm 3: Diagnosis of Cellular and Combined Immunodeficiencies
Algorithm 4: Diagnosis of Phagocyte Defects
Algorithm 5: Diagnosis of Complement Deficiency
Algorithm 6: General Considerations for Therapy of Primary Immunodeficiency
Classification of primary immunodeficiencies: Need for a revised approach? http://buff.ly/YBkwI7
Published: 08/29/2009
Updated: 03/02/2014
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
Since Bruton described X-linked agammaglobulinemia in 1952, more than 180 different primary immunodeficiencies have been described.
10 Warning Signs of Primary Immunodeficiency in Children
1. Four or more new ear infections within one year.
2. Two or more sinus infections within one year, or chronic sinusitis.
3. Two or more months on antibiotics with little effect.
4. Two or more pneumonias within one year, or pneumonia twice over any time.
5. Failure of an infant to gain weight or grow normally.
6. Recurrent, deep skin or organ abscesses.
7. Persistent thrush in mouth or fungal infection on skin.
8. Need for intravenous antibiotics to clear infection.
9. Two or more deep-seated infections including septicemia.
10. A family history of primary immunodeficiency.
10 Warning Signs of Primary Immunodeficiency in Adults
1. Two or more new ear infections within one year.
2. Two or more serious sinus infections within one year, in the absence of an allergy; or chronic sinusitis.
3. One pneumonia per year for more than one year, or pneumonia twice over any time.
4. Chronic diarrhea with weight loss.
5. Recurrent viral infections (colds, herpes, warts, condyloma).
6. Recurrent need for intravenous antibiotics to clear infections.
7. Recurrent, deep abscesses of the skin or internal organs.
8. Persistent thrush or fungal infection on skin or elsewhere.
9. Infection with normally harmless tuberculosis-like bacteria.
10. A family history of primary immunodeficiency.
Source: JMFworld.com. PDF handouts: Children PIDD, Children PIDD (illustrated), Adult PIDD, 4 Stages of Immunologic Testing when PIDD is suspected.
Primary immunodeficiency disorders (PIDD) (click to enlarge the image).
Definition of PIDD: Genetically determined immunodeficiency.
The World Health Organization recognizes more than 100 PIDD. Most of PIDD are rare but selective IgA deficiency is relatively common, prevalence 1:500.
4 Major Host Defense Mechanisms:
- B-cell immunity
- T-cell immunity
- Phagocytic cells
- Complement system
Phagocyte immunodeficiencies (click to enlarge the image):
Read more:
Phagocyte Deficiencies
Chronic Granulomatous Disease (CGD)
Chediak-Higashi Syndrome (CHS)
Leukocyte adhesion deficiency (LAD)
Leukocyte adhesion deficiency type I (LAD I)
Leukocyte adhesion deficiency type II (LAD II)
Leukocyte adhesion deficiency type III (LAD III)
Hyper IgE Syndrome (HIES)

Humoral immunodeficiency (click to enlarge the image).
B and T cells "talk" constantly in T-dependent immune responses where a host of enzymes are involved in class switching - AICD, UNG, CD40, CD40L - if there is a defect in any of these enzymes, B cells cannot class switch - Ig gets "stuck" at IgM level and hyper-IgM immonodeficiency develops.

T-cell Immunodeficiencies (click to enlarge the image).
The autoimmune regulatory gene (AIRE) is expressed in the thymus. AIRE promotes expression of non-thymic tissue antigens in the thymus - a key part of the thymic education of T cells.
Mutation in the AIRE gene produces disorders such as autoimmune polyglandular syndrome or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). In this syndrome, T cells do not develop tolerance to self-antigens. The endocrine organs are attacked by autoreactive T cells. The AIRE mutation was the first report of a single-gene defect causing a systemic human autoimmune disease.
Mutations in FAS or caspase 10 manifest as autoimmune lymphoproliferative syndrome (ALPS). In ALPS, lymphocytes do not get a signal to "die" and they accumulate in the lymphoid organs.
Read more:
IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X linked) syndrome
Autoimmune lymphoproliferative syndrome (ALPS)
Chronic Mucocutaneous Candidiasis (CMCC)
Diagnosis of T-cell Immunodeficiency

Combined Immunodeficiencies (click to enlarge the image).
Bare lymphocyte syndromes include MHC class I and MHC class II deficiencies. These are primary immune deficiency disorders (PIDD) due to a lack of expression of either MHC I or MHC II. MHC class I deficiency leads to CD8 lymphopenia. MHC class II deficiency leads to CD4 lymphopenia.
Read more:
DiGeorge Syndrome (DGS)
Wiskott-Aldrich Syndrome (WAS)
Ataxia-Telangiectasia (A-T)
.jpg)
Severe Combined Immunodeficiency (SCID) (click to enlarge the image).
In SCID, the younger the age of the patient at the time of transplantation, the better the prognosis. There is a 95% survival rate in an infant who undergoes a transplant before 3 months of age. After six months, the survival rate decreases dramatically, to 50%.
Receptor editing reactivates RAG-1 and RAG-2 when a high affinity self-antigen is recognized by a B cell receptor (BCR). RAG-1 and RAG-2 defects lead to Omenn syndrome, a form of SCID.
Severe combined immunodeficiency (SCID) - 4 groups according to T/B/NK cells (click to enlarge the image).
Receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 contain γ chain, which is affected in X-linked SCID.
Cytokine receptors (click to enlarge the image)

Complement deficiencies (click to enlarge the image).
Read more in Complement Deficiencies.
Tests and Workup of Suspected Immunodeficiency - mostly primary immunodeficiency disorders (PIDD)
B-cell PIDD
Flow Cytometry: CD19 and CD20
Ig G, A, M, E
IgG subclasses (IgG1, IgG2, IgG3, IgG4)
IgG titer for Tetanus
IgG titer for Diphtheria
IgG titer for Haemophilus influenzae type B
IgG titer for Mumps
IgG titers for Pneumococcus
IgG titers for Pneumococcal conjugated vaccine (if given PCV7 or PCV13, Prevnar in the U.S.)
IgG titers for Pneumococcal unconjugated vaccine (if given PSPV, Pneumovax in the U.S.)
Blood group Isohemagglutinins (for diagnosis of CVID)
Mitogen stimulation assays for B- and T-cells
T-cell PIDD (T-cell and NK cell)
Absolute lymphocyte count (CBC-Diff)
Flow Cytometry: CD3CD4, CD3CD8, and CD16CD56
Delayed-type hypersensitivity
Enzyme assays (ADA and PNP)
NK cytolysis assay
Mitogen stimulation assays for B- and T-cells
Phagocyte PIDD (neutrophils, macrophages and monocytes)
CBC-Diff with peripheral smear
Absolute neutrophil count
Flow Cytometry: LFA-1 (CD11a or CD18) and CD15
Oxidative function (DHR or NBT or chemiluminescence)
Enzyme assays (MPO and G6PDH)
Phagocyte function assay - chemotaxis and bactericidal function
Complement PIDD
C3 and C4
C1q
C1 esterase inhibitor (C1-INH) - qualitative and quantitative
CH50 (classic pathway)
AH50 (alternative pathway)
References
Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol 2005 May;94(5 Suppl 1):S1-63.
Algorithm 1: General Approach for the Diagnosis of Primary Immunodeficiency
Algorithm 2: Diagnosis of Humoral Immunodeficiency
Algorithm 3: Diagnosis of Cellular and Combined Immunodeficiencies
Algorithm 4: Diagnosis of Phagocyte Defects
Algorithm 5: Diagnosis of Complement Deficiency
Algorithm 6: General Considerations for Therapy of Primary Immunodeficiency
Classification of primary immunodeficiencies: Need for a revised approach? http://buff.ly/YBkwI7
Published: 08/29/2009
Updated: 03/02/2014
How to Diagnose Common Variable Immunodeficiency (CVID)?
Author: V. Dimov, M.D., Allergist/Immunologist and Assistant Professor at University of Chicago
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
A 65-year-old Caucasian female (CF) is referred by her hematologist and primary care physician (PCP) for work-up of suspected immunodeficiency. She was referred to hematology by her PCP for work-up of frequent urinary tract infections (UTIs) for the last year. Complete blood count with differential (CBCD), bone marrow and chromosomal analysis were negative. Serum immunoglobulins showed low levels of IgG 515 mg/dL (reference range 700 to 1600), IgM 30 (reference range 40 to 230) , IgA 50 (reference range 70 to 400). UA: bacteriuria, no protein. ANA was negative.
Physical examination
Normal.
What is the reason for the low levels of immunoglobulins?
Differential diagnosis:
1. Immunoglobulin loss from GI tract, for example, protein-losing enteropathy. These patients usually have diarrhea but our patient complains of constipation.
2. Immunoglobulin loss in the urine, for example, nephrotic syndrome. IgM (pentamer) and IgA (dimer) are larger molecules than IgG. It is very unlikely to see low levels for IgM in patients with nephrotic syndrome or protein-losing enteropathy. IgG gets filtered in the urine but the large IgM pentamer does not. A 24-hour urine collection is the best method to determine the amount of protein in the urine.
3. Low production of Ig due to bone marrow process or CLL. Both were excluded by BM biopsy and normal CBCD.
4. Common variable immunodeficiency (CVID)
Common variable immunodeficiency (CVID) is the most likely diagnosis in this patient.
How do you diagnose CVID?
A patient with suspected CVID can be tested for the ability to mount humoral and cellular immune response as follows:
1. Humoral immune response.
Inject patient with Pneumovax 23 (not Prevnar, which is a 7-valent conjugate vaccine). Check anti-polysaccharide IgG antibody to pneumococcus serotypes in 3 weeks. There should be a 3-5 fold increase in the anitbody titer to at least 50% of isotypes.
Which serotypes should be included in the order for Ig?
The same serotypes that were included in the given vaccine. Check the enclosed leaflet for this information. For example, the serotypes included in Pneumovax 23 can be found from the Merck website (PDF):
1 2 3 4 5 6B 7F 8 9N 9V 10A 11A 12F 14 15B 17F 18C 19F 19A 20 22F 23F 33F
How to collect the serum for anti-polysaccharide IgG antibodies?
Collect the serum prior to immunization with Pneumovax 23. Store the pre-immunization serum in the office refrigerator. Give the vaccine. Check the post-immunization serum 3 weeks later. Send both pre- and post-immunization serums to the laboratory at the same time.
The pneumococcal vaccine comprises purified capsular polysaccharide of 23 stereotypes that account for more than 90% of the invasive pneumococcal infections in the USA. It induces anti-polysaccharide IgG antibody levels to most or all of the component polysaccharide antigens in immunocompetent adults. Elderly adults respond equally well to vaccination as do younger adults. The current 23-valent vaccine comprises 25 μg of each of 23 pneumococcal stereotypes (namely, serotypes 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F and 33F).
The test should be ordered as follows: Pre- and post-IgM and IgG antibody titers to pneumococcal serotypes 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F and 33F
2. Cellular immune response.
Some patients with CVID have decreased cellular immunity as well. Cellular immunity is tested by anergy panel with common antigens to which the patient was likely exposed in the past: Tetanus toxoid, Trichophyton, Candida. An intradermal injection of 0.1 mL of each antigen is necessary to perform the test. The test is read in 48-72 hours. A positive test result indicates intact delayed-type hypersensitivity. A negative test result to all antigens suggests impaired type IV immunity.
Flow cytometry should also be ordered. Lymphocyte proliferation is a more sensitive test to evaluate cellular immune response.
Delayed-type hypersensitivity (DTH) response
The standardized DTH test includes Candida, tetanus, mumps, and TB. Trichophyton is also commonly used. However, the only FDA-approved reagents for DTH are PPD, Candida and mumps.
What are the normal serum immunoglobulin levels (IgG, IgA, IgM)?
Serum levels of IgM, IgG and IgA vary with age, gender and race.
The IgG and IgA concentrations in children show a gradual rise with increasing age. The IgA level is generally about the same in both sexes. Girls typically have higher IgM and IgG levels than boys.
The confidence interval bounded by two standard deviations about the mean excludes 5% of apparently healthy controls.
Elevated IgM, low IgA, low IgG, low IgM, and elevated IgA are the commonest changes observed in apparently healthy humans.
Humoral immunodeficiency is commonly defined as IgG, IgM or IgA level that is two standard deviations (2 SD) below the mean level for IgG, IgM or IgA, respectively, for the particular age group and gender.
Serum levels of IgM, IgG and IgA. Source: Pediatrics, 1966 and Immunologic disorders in infants and children, by E. Richard Stiehm, Hans D. Ochs, Jerry A. Winkelstein.
Typical pattern of immunoglobulin levels (IgG, IgA, IgM) in humoral immunodeficiency. Click here to enlarge the table.
References
Diagnostic Criteria for Common Variable Immunodeficiency (CVID): Probable and Possible Diagnosis
The diagnostic criteria are divided into three categories: definitive, probable, and possible. There are no criteria for definitive diagnosis of Common Variable Immunodeficiency (CVID) at this time.
To guard against the inclusion of patients who have polymorphic variants in the genes associated with immunodeficiency and to specify the clinical or laboratory finding that is most consistently abnormal in a particular disorder, the patient must fulfill an inclusion criterion that is characteristic of the disorder.
Definitive diagnosis
Patients with a definitive diagnosis are assumed to have a greater than 98% probability that in 20 years they will still be given the same diagnosis. Mutation detection is the most reliable method of making a diagnosis but a single mutation is rarely found in CVID.
Probable diagnosis
Patients with a probable diagnosis are those with all of the clinical and laboratory characteristics of a particular disorder but who do not have a documented abnormality in the gene, the mRNA, or the protein that is known to be abnormal in the disorder. They are assumed to have a greater than 85% probability that in 20 years they will be given the same diagnosis.
Probable diagnosis of CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in serum IgG AND IgA and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Possible diagnosis
Patients with a possible diagnosis are those that have some but not all of the characteristic clinical or laboratory findings of a particular disorder.
Possible diagnosis if CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in one of the major isotypes (IgM, IgG, and IgA) and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Clinical features of CVID
Most patients with CVID are diagnosed with immunodeficiency in the second, third, or fourth decade of life, after they have had several pneumonias; however, children and older adults may be affected.
Viral, fungal, and parasitic infections as well as bacterial infections may be found.
The serum concentration of IgM is normal in about half of the patients.
Abnormalities in T cell numbers or function are common. The majority of patients have normal numbers of B cells; however, some have low or absent B cells.
Approximately 50% of patients have autoimmune manifestations. There is an increased risk of malignancy.
Differential diagnosis of hypogammaglobulinemia includes drug-induced, for example secondary to glucocorticoids (steroids).
Diagnostic Criteria for Primary Immunodeficiencies. Mary Ellen Conley, Luigi D. Notarangelo, and Amos Etzioni Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clinical Immunology, Vol. 93, No. 3, December, pp. 190–197, 1999.
What is the treatment for CVID?
Intravenous immunoglobulin (IVIG) administered at outpatient clinic, monthly, this is the most common treatment.
Subcutaneous immunoglobulin G (SCIG), administered at home, given weekly.
Summary
CVID is characterized by:
1. Low levels of most or all of the immunoglobulin (Ig) classes
2. Lack of B lymphocytes or plasma cells that are capable of producing antibodies
3. Frequent bacterial infections
CVID is the most common primary immunodeficiency with an incidence of 1 case per 10,000-50,000 population. More than two thirds of patients are aged 21 years or older when CVID is diagnosed. CVID represents a group of heterogeneous conditions. Approximately 50% of patients with the deficiency also have diminished serum immunoglobulin M (IgM) levels and T-lymphocyte dysfunction. About 20% of those with CVID have an autoimmune disease.
The prognosis for patients with CVID is good if they do not have bronchiectasis, autoimmune disease or malignancy.

Five immunoglobulin classes (mind map)
In order of their serum concentrations:
IgG 1000 mg/dL
IgA 200 mg/dL
IgM 150 mg/dL
IgD 4 mg/dL
IgE 0.005 mg/dL (extremely low serum concentration compared to other Ig in (GAMED)
IgG and A are divided in subclasses: 4 for IgG -- IgG1, IgG2, IgG3, IgG4, and 2 for IgA -- IgA1 and IgA2.

Ig structures. Image source: Wikipedia.
Can patients with CVID receive live vaccines?
Patients with CVID should never receive live vaccines and should be advised to avoid contact with children or adults who have recently been given a live vaccine, for example, FluMist® nasal flu vaccine spray.
Can patients with CVID travel to endemic area for hepatitis A or B?
Yes but they should be given hyperimmune Ig prior to travel to endemic areas. The hyperimmune Ig provides protection for 3 weeks.
What is the half-life of IVIG?
3 weeks.
Mnemonic: Dose of IVIG in PIDD
400-600 mg/kg/month
4 letter words:
IVIG
CVID
SCID
What are the long-term risks for patients with CVID?
Malignancies, autoimmune diseases, infections.
FDA guidelines for IVIG state the product should be prepared out of at least 1,000 different human donors. Most commercial IVIG preparations contain products from over 10,000 human donors which increases the risk of acquiring diseases such as HIV, HCV or Creutzfeldt-Jakob disease. The risk is small but nevertheless, it should be discussed with the patient.
What HIV screening test would you do in a patient with CVID?
Patients with CVID can not produce an adequate antibody response to the HIV virus if infected and ELISA or Western blot are not useful. PCR RNA is the test of choise for HIV diagnosis in patients with CVID.
Can patients with CVID still have allergies?
Yes, the production of IgE may not be affected in patients with CVID can have symtpoms of allergic rhinitis, urticaria and other IgE-mediated diseases.
Differences between commercial IVIG preparations: Gammagard and Octagam
Gammagard Liquid is the first 10% IVIG solution with no added carbohydrates or sodium. Baxter is replacing Gammagard S/D with Gammagard Liquid.
Sodium concentration
Gammagard: No added sodium. When reconstituted with the total volume of diluent (Sterile Water for Injection, USP) supplied to 5%, Gammagard contains a physiological concentration of sodium chloride (approximately 8.5 mg/mL) and has a pH of 6.8 ± 0.4.
Octagam: sodium content of the final solution is ≤ 30 mmol/l and the pH is between 5.1 and 6.0.
Osmolality
Gammagard osmolality is 240-300 mOsmol/kg, similar to physiological osmolality (285 to 295 mOsmol/kg).
Octagam osmolality is 310 - 380 mosmol/kg.
Cost
Gammagard Liquid, Baxter, $129.60/gm
Octagam, Octapharma, $118.34/gm
References
Antibody response of pneumococcal vaccine: need for booster dosing? International Journal of Antimicrobial Agents, Volume 14, Issue 2, March 2000, Pages 107-112.
How to identify a possible specific antibody deficiency to pneumococcus. AAAAI, 2007.
Common Variable Immunodeficiency. eMedicine.
Common Variable Immunodeficiency. eMedicine.
Common Variable Immunodeficiency: A Multifaceted And Puzzling Disorder. Expert Review of Clinical Immunology, Medscape, 2009.
Published: 07/12/2008
Updated: 02/06/2012
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
A 65-year-old Caucasian female (CF) is referred by her hematologist and primary care physician (PCP) for work-up of suspected immunodeficiency. She was referred to hematology by her PCP for work-up of frequent urinary tract infections (UTIs) for the last year. Complete blood count with differential (CBCD), bone marrow and chromosomal analysis were negative. Serum immunoglobulins showed low levels of IgG 515 mg/dL (reference range 700 to 1600), IgM 30 (reference range 40 to 230) , IgA 50 (reference range 70 to 400). UA: bacteriuria, no protein. ANA was negative.
Physical examination
Normal.
What is the reason for the low levels of immunoglobulins?
Differential diagnosis:
1. Immunoglobulin loss from GI tract, for example, protein-losing enteropathy. These patients usually have diarrhea but our patient complains of constipation.
2. Immunoglobulin loss in the urine, for example, nephrotic syndrome. IgM (pentamer) and IgA (dimer) are larger molecules than IgG. It is very unlikely to see low levels for IgM in patients with nephrotic syndrome or protein-losing enteropathy. IgG gets filtered in the urine but the large IgM pentamer does not. A 24-hour urine collection is the best method to determine the amount of protein in the urine.
3. Low production of Ig due to bone marrow process or CLL. Both were excluded by BM biopsy and normal CBCD.
4. Common variable immunodeficiency (CVID)
Common variable immunodeficiency (CVID) is the most likely diagnosis in this patient.
How do you diagnose CVID?
A patient with suspected CVID can be tested for the ability to mount humoral and cellular immune response as follows:
1. Humoral immune response.
Inject patient with Pneumovax 23 (not Prevnar, which is a 7-valent conjugate vaccine). Check anti-polysaccharide IgG antibody to pneumococcus serotypes in 3 weeks. There should be a 3-5 fold increase in the anitbody titer to at least 50% of isotypes.
Which serotypes should be included in the order for Ig?
The same serotypes that were included in the given vaccine. Check the enclosed leaflet for this information. For example, the serotypes included in Pneumovax 23 can be found from the Merck website (PDF):
1 2 3 4 5 6B 7F 8 9N 9V 10A 11A 12F 14 15B 17F 18C 19F 19A 20 22F 23F 33F
How to collect the serum for anti-polysaccharide IgG antibodies?
Collect the serum prior to immunization with Pneumovax 23. Store the pre-immunization serum in the office refrigerator. Give the vaccine. Check the post-immunization serum 3 weeks later. Send both pre- and post-immunization serums to the laboratory at the same time.
The pneumococcal vaccine comprises purified capsular polysaccharide of 23 stereotypes that account for more than 90% of the invasive pneumococcal infections in the USA. It induces anti-polysaccharide IgG antibody levels to most or all of the component polysaccharide antigens in immunocompetent adults. Elderly adults respond equally well to vaccination as do younger adults. The current 23-valent vaccine comprises 25 μg of each of 23 pneumococcal stereotypes (namely, serotypes 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F and 33F).
The test should be ordered as follows: Pre- and post-IgM and IgG antibody titers to pneumococcal serotypes 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F and 33F
2. Cellular immune response.
Some patients with CVID have decreased cellular immunity as well. Cellular immunity is tested by anergy panel with common antigens to which the patient was likely exposed in the past: Tetanus toxoid, Trichophyton, Candida. An intradermal injection of 0.1 mL of each antigen is necessary to perform the test. The test is read in 48-72 hours. A positive test result indicates intact delayed-type hypersensitivity. A negative test result to all antigens suggests impaired type IV immunity.
Flow cytometry should also be ordered. Lymphocyte proliferation is a more sensitive test to evaluate cellular immune response.
Delayed-type hypersensitivity (DTH) response
The standardized DTH test includes Candida, tetanus, mumps, and TB. Trichophyton is also commonly used. However, the only FDA-approved reagents for DTH are PPD, Candida and mumps.
What are the normal serum immunoglobulin levels (IgG, IgA, IgM)?
Serum levels of IgM, IgG and IgA vary with age, gender and race.
The IgG and IgA concentrations in children show a gradual rise with increasing age. The IgA level is generally about the same in both sexes. Girls typically have higher IgM and IgG levels than boys.
The confidence interval bounded by two standard deviations about the mean excludes 5% of apparently healthy controls.
Elevated IgM, low IgA, low IgG, low IgM, and elevated IgA are the commonest changes observed in apparently healthy humans.
Humoral immunodeficiency is commonly defined as IgG, IgM or IgA level that is two standard deviations (2 SD) below the mean level for IgG, IgM or IgA, respectively, for the particular age group and gender.
Serum levels of IgM, IgG and IgA. Source: Pediatrics, 1966 and Immunologic disorders in infants and children, by E. Richard Stiehm, Hans D. Ochs, Jerry A. Winkelstein.
Typical pattern of immunoglobulin levels (IgG, IgA, IgM) in humoral immunodeficiency. Click here to enlarge the table.
References
Serum immunoglobulin levels in healthy children and adults. J. W. Stoop, B. J. M. Zegers, P. C. Sander, and R. E. Ballieux. Clin Exp Immunol. 1969 January; 4(1): 101–112.
The relationship of race, sex, and age to concentrations of serum immunoglobulins expressed in international units in healthy adults in the USA. S. E. Maddison, C. C. Stewart, C. E. Farshy, and C. B. Reimer. Bull World Health Organ. 1975; 52(2): 179–185.
Serum immunoglobulin concentrations in preschool children measured by laser nephelometry: reference ranges for IgG, IgA, IgM. D Isaacs, D G Altman, C E Tidmarsh, H B Valman, and A D Webster. J Clin Pathol. 1983 October; 36(10): 1193–1196.
D Isaacs, A D Webster, and H B Valman. Clin Exp Immunol. 1984 November; 58(2): 335–340.
Serum Immunoglobulin Levels Throughout the Life-Span of Healthy Man. Ann of Int Med, November 1, 1971, Vol. 75 no. 5 673-682.
Diagnostic Criteria for Common Variable Immunodeficiency (CVID): Probable and Possible Diagnosis
The diagnostic criteria are divided into three categories: definitive, probable, and possible. There are no criteria for definitive diagnosis of Common Variable Immunodeficiency (CVID) at this time.
To guard against the inclusion of patients who have polymorphic variants in the genes associated with immunodeficiency and to specify the clinical or laboratory finding that is most consistently abnormal in a particular disorder, the patient must fulfill an inclusion criterion that is characteristic of the disorder.
Definitive diagnosis
Patients with a definitive diagnosis are assumed to have a greater than 98% probability that in 20 years they will still be given the same diagnosis. Mutation detection is the most reliable method of making a diagnosis but a single mutation is rarely found in CVID.
Probable diagnosis
Patients with a probable diagnosis are those with all of the clinical and laboratory characteristics of a particular disorder but who do not have a documented abnormality in the gene, the mRNA, or the protein that is known to be abnormal in the disorder. They are assumed to have a greater than 85% probability that in 20 years they will be given the same diagnosis.
Probable diagnosis of CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in serum IgG AND IgA and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Possible diagnosis
Patients with a possible diagnosis are those that have some but not all of the characteristic clinical or laboratory findings of a particular disorder.
Possible diagnosis if CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in one of the major isotypes (IgM, IgG, and IgA) and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Clinical features of CVID
Most patients with CVID are diagnosed with immunodeficiency in the second, third, or fourth decade of life, after they have had several pneumonias; however, children and older adults may be affected.
Viral, fungal, and parasitic infections as well as bacterial infections may be found.
The serum concentration of IgM is normal in about half of the patients.
Abnormalities in T cell numbers or function are common. The majority of patients have normal numbers of B cells; however, some have low or absent B cells.
Approximately 50% of patients have autoimmune manifestations. There is an increased risk of malignancy.
Differential diagnosis of hypogammaglobulinemia includes drug-induced, for example secondary to glucocorticoids (steroids).
References
Diagnostic Criteria for Primary Immunodeficiencies. Mary Ellen Conley, Luigi D. Notarangelo, and Amos Etzioni Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clinical Immunology, Vol. 93, No. 3, December, pp. 190–197, 1999.
Recognizing Primary Immune Deficiency in Clinical Practice. Clinical and Vaccine Immunology, March 2006, p. 329-332, Vol. 13, No. 3.
What is the treatment for CVID?
Intravenous immunoglobulin (IVIG) administered at outpatient clinic, monthly, this is the most common treatment.
Subcutaneous immunoglobulin G (SCIG), administered at home, given weekly.
Summary
CVID is characterized by:
1. Low levels of most or all of the immunoglobulin (Ig) classes
2. Lack of B lymphocytes or plasma cells that are capable of producing antibodies
3. Frequent bacterial infections
CVID is the most common primary immunodeficiency with an incidence of 1 case per 10,000-50,000 population. More than two thirds of patients are aged 21 years or older when CVID is diagnosed. CVID represents a group of heterogeneous conditions. Approximately 50% of patients with the deficiency also have diminished serum immunoglobulin M (IgM) levels and T-lymphocyte dysfunction. About 20% of those with CVID have an autoimmune disease.
The prognosis for patients with CVID is good if they do not have bronchiectasis, autoimmune disease or malignancy.

Five immunoglobulin classes (mind map)
In order of their serum concentrations:
IgG 1000 mg/dL
IgA 200 mg/dL
IgM 150 mg/dL
IgD 4 mg/dL
IgE 0.005 mg/dL (extremely low serum concentration compared to other Ig in (GAMED)
IgG and A are divided in subclasses: 4 for IgG -- IgG1, IgG2, IgG3, IgG4, and 2 for IgA -- IgA1 and IgA2.

Ig structures. Image source: Wikipedia.
{kind=link}
Can patients with CVID receive live vaccines?
Patients with CVID should never receive live vaccines and should be advised to avoid contact with children or adults who have recently been given a live vaccine, for example, FluMist® nasal flu vaccine spray.
Can patients with CVID travel to endemic area for hepatitis A or B?
Yes but they should be given hyperimmune Ig prior to travel to endemic areas. The hyperimmune Ig provides protection for 3 weeks.
What is the half-life of IVIG?
3 weeks.
Mnemonic: Dose of IVIG in PIDD
400-600 mg/kg/month
4 letter words:
IVIG
CVID
SCID
What are the long-term risks for patients with CVID?
Malignancies, autoimmune diseases, infections.
FDA guidelines for IVIG state the product should be prepared out of at least 1,000 different human donors. Most commercial IVIG preparations contain products from over 10,000 human donors which increases the risk of acquiring diseases such as HIV, HCV or Creutzfeldt-Jakob disease. The risk is small but nevertheless, it should be discussed with the patient.
What HIV screening test would you do in a patient with CVID?
Patients with CVID can not produce an adequate antibody response to the HIV virus if infected and ELISA or Western blot are not useful. PCR RNA is the test of choise for HIV diagnosis in patients with CVID.
Can patients with CVID still have allergies?
Yes, the production of IgE may not be affected in patients with CVID can have symtpoms of allergic rhinitis, urticaria and other IgE-mediated diseases.
Differences between commercial IVIG preparations: Gammagard and Octagam
Gammagard Liquid is the first 10% IVIG solution with no added carbohydrates or sodium. Baxter is replacing Gammagard S/D with Gammagard Liquid.
Sodium concentration
Gammagard: No added sodium. When reconstituted with the total volume of diluent (Sterile Water for Injection, USP) supplied to 5%, Gammagard contains a physiological concentration of sodium chloride (approximately 8.5 mg/mL) and has a pH of 6.8 ± 0.4.
Octagam: sodium content of the final solution is ≤ 30 mmol/l and the pH is between 5.1 and 6.0.
Osmolality
Gammagard osmolality is 240-300 mOsmol/kg, similar to physiological osmolality (285 to 295 mOsmol/kg).
Octagam osmolality is 310 - 380 mosmol/kg.
Cost
Gammagard Liquid, Baxter, $129.60/gm
Octagam, Octapharma, $118.34/gm
References
Antibody response of pneumococcal vaccine: need for booster dosing? International Journal of Antimicrobial Agents, Volume 14, Issue 2, March 2000, Pages 107-112.
How to identify a possible specific antibody deficiency to pneumococcus. AAAAI, 2007.
Common Variable Immunodeficiency. eMedicine.
Common Variable Immunodeficiency. eMedicine.
Common Variable Immunodeficiency: A Multifaceted And Puzzling Disorder. Expert Review of Clinical Immunology, Medscape, 2009.
Primary Immunodeficiency Diseases: Definition, Diagnosis, and Management. Nima Rezaei, Luigi D. Notarangelo, Asghar Aghamohammadi, 2008, Google eBook.
Common variable immunodeficiency (CVID). Allergologia et Immunopathologia. November 2006. Volume 34 - Number 06 p. 263 - 275.
Adaptive Humoral Immunity: B-cells and Immunoglobulins
Mnemonics: Adaptive Humoral Immunity: B-cells and Immunoglobulins
Intravenous immune globulin. Hopkins HIV Guide.
Common variable immunodeficiency (CVID). Allergologia et Immunopathologia. November 2006. Volume 34 - Number 06 p. 263 - 275.
Adaptive Humoral Immunity: B-cells and Immunoglobulins
Mnemonics: Adaptive Humoral Immunity: B-cells and Immunoglobulins
Intravenous immune globulin. Hopkins HIV Guide.
Evaluation and diagnosis of common variable immunodeficiency. UpToDate.
Octagam, Octapharma.
Gammagard Liquid, Baxter.
Octagam, Octapharma.
Gammagard Liquid, Baxter.
Interpretation of pneumococcal antibody titers - AAAAI Ask the Expert, 2011.
Assessment of antibody response to pneumococcus immunization. AAAAI, Ask the Expert, 2011.
Cost-effectiveness of Pneumococcal Conjugate Vaccine vs. Polysaccharide Vaccine in Adults: PCV13 was better than PPSV23. JAMA, 2012.
Cost-effectiveness of Pneumococcal Conjugate Vaccine vs. Polysaccharide Vaccine in Adults: PCV13 was better than PPSV23. JAMA, 2012.
Published: 07/12/2008
Updated: 02/06/2012
Antibody titer responses to pneumococcal vaccination in common variable immunodeficiency (CVID)
Author: V. Dimov, M.D., Allergist/Immunologist and Assistant Professor at University of Chicago
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
A 54-year-old Caucasian male (CM) is evaluated by the Allergy and Immunology clinic for recurrent sinusitis for the last 20-30 years with 5-6 episodes per year. His IgG level was 470 mg/dL two months ago and IgM and IgA levels were normal. He reports no other significant infections. The patient reports seasonal exacerbations of his nasal symptoms in the spring, fall and winter. His last sinus infection was 9 months ago and he has not had such a recurrence free period in years.
Past medical history
Hypertension (HTN), hyperlipidemia (HLP), sinusitis, non-allergic rhinitis (non-AR), dermatographism.
Medications
Fexofenadine, flunisolide nasal spray, lisinopril.
Physical examination
Normal.
What is the most likely diagnosis?
Common Variable Immunodeficiency (CVID).
Allergic or non-allergic rhinitis.
What tests would you order?
Pre- and post-pneumococcal immunization titers.
ANA, HIV, RF.
B and T-cell flow cytometry.
RAST to aeroallergens.
What happened?
Skin prick testing: uninterpretable due to dermatographism. RAST to aeroallergens: negative.
HIV, ANA, RF were negative. CXR and U/A without evidence of malignancy. He had a colonoscopy in the past 3 years and it was negative.


Pre-immunization titers (left). Post-immunization titers (right) (click to enlarge the images).
Pre- and post-pneumococcal immunization titers 4 weeks apart showed an insufficient increase in the antibody titers: only 30% (4 out of 13 titers) of specific antibodies increased by the magnitude of 2 fold.
Flow Cytometry. Specimen: Peripheral Blood. Processing: Lyse, cytospin, stain with monoclonal antibodies. Cell Count: 8.2 10*6 total cells. Viability: 89%.
Total events analyzed = 42415
A gate Bright CD45 positive events = 5758 (17% of total events analyzed)
Interpretation: Lymphocytes comprise 17% of the total events analyzed, with 3% B-cells, 85% T-cells, and 11% NK cells. The CD4:CD8 ratio is 2.7:1. 9% of the lymphocytes are CD25 (dim) positive T-cells.
In healthy subjects, B cells constitute 18-47% of the peripheral lymphocytes. B cell count can be either low or normal in patients with CVID.
What happened next?
The patient was diagnosed with common variable immunodeficiency (CVID).
Would you start intravenous immunoglobulin G (IVIG) therapy for this patient?
Historically, the level of IgG of 470 mg/dL did not typically require replacement with IVIG in the absence of severe recurrent infections. Some of the newer studies point to an optimal replacement level of IgG of 600-800 mg/dL.
We decided to repeat the quantitative serum immunoglobulins in 4-6 months and to decide about IVIG replacement at that point.
Final diagnosis
Common Variable Immunodeficiency (CVID).
What did we learn from this case?
Evaluation of the response to pneumococcal vaccination is best accomplished by comparing pre-vaccination and post-vaccination antibody levels. A 2 to 4 fold increase in type-specific antibodies measured 4-6 weeks after vaccination is expected in immunocompetent adults. The number of serotypes for which a 2 to 4 fold increase is observed varies greatly among individuals; a consensus panel has suggested that individuals older than 5 years should respond to at least approximately 70% of pneumococcal serotypes. Adults older than 65 years may exhibit a smaller (less than 2 fold) increase in type specific antibody levels.
The "rule of thumb" is that 2/3 of titers have to increase 2-3 fold.
The patient did not have evidence of allergen sensitization on ImmunoCAP test for aeroallergens, therefore the intranasal steroids and oral antihistamine were discontinued. He was advised to use nasal saline rinses BID.
What are the normal serum immunoglobulin levels (IgG, IgA, IgM)?
Serum levels of IgM, IgG and IgA vary with age, gender and race.
The IgG and IgA concentrations in children show a gradual rise with increasing age. The IgA level is generally about the same in both sexes. Girls typically have higher IgM and IgG levels than boys.
The confidence interval bounded by two standard deviations about the mean excludes 5% of apparently healthy controls.
Elevated IgM, low IgA, low IgG, low IgM, and elevated IgA are the commonest changes observed in apparently healthy humans.
Humoral immunodeficiency is commonly defined as IgG, IgM or IgA level that is two standard deviations (2 SD) below the mean level for IgG, IgM or IgA, respectively, for the particular age group and gender.
Serum levels of IgM, IgG and IgA. Source: Pediatrics, 1966 and Immunologic disorders in infants and children, by E. Richard Stiehm, Hans D. Ochs, Jerry A. Winkelstein.
Typical pattern of immunoglobulin levels (IgG, IgA, IgM) in humoral immunodeficiency. Click here to enlarge the table.
What is a normal response to pneumococcal immunization?
Protection against infection and colonization is associated with specific IgG concentrations of 1.3 mcg/ml.
A second component in judging response is the final concentration of antibodies after immunization regardless of increase from preimmunization concentration. An adequate response is a post-immunization antibody concentration of more than 1.3 mcg/ml, even in the absence of a fourfold increase.
Children 2 to 5 years of age are normally expected to have an adequate response to more than 50% of serotypes tested. Patients 6 years or older are normally expected to respond to more than 70% of the serotypes tested. Source: Assessment and clinical interpretation of polysaccharide antibody responses. Kenneth Paris and Ricardo Sorensen. Annals of Allergy, Asthma, and Immunology, 2007; volume 99, Issue 5, Pages 462-464, and AAAAI Ask the Expert, 2012.
References
Diagnostic Criteria for Common Variable Immunodeficiency (CVID): Probable and Possible Diagnosis
The diagnostic criteria are divided into three categories: definitive, probable, and possible. There are no criteria for definitive diagnosis of Common Variable Immunodeficiency (CVID) at this time.
To guard against the inclusion of patients who have polymorphic variants in the genes associated with immunodeficiency and to specify the clinical or laboratory finding that is most consistently abnormal in a particular disorder, the patient must fulfill an inclusion criterion that is characteristic of the disorder.
Definitive diagnosis
Patients with a definitive diagnosis are assumed to have a greater than 98% probability that in 20 years they will still be given the same diagnosis. Mutation detection is the most reliable method of making a diagnosis but a single mutation is rarely found in CVID.
Probable diagnosis
Patients with a probable diagnosis are those with all of the clinical and laboratory characteristics of a particular disorder but who do not have a documented abnormality in the gene, the mRNA, or the protein that is known to be abnormal in the disorder. They are assumed to have a greater than 85% probability that in 20 years they will be given the same diagnosis.
Probable diagnosis of CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in serum IgG AND IgA and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Possible diagnosis
Patients with a possible diagnosis are those that have some but not all of the characteristic clinical or laboratory findings of a particular disorder.
Possible diagnosis if CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in one of the major isotypes (IgM, IgG, and IgA) and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Clinical features of CVID
Most patients with CVID are diagnosed with immunodeficiency in the second, third, or fourth decade of life, after they have had several pneumonias; however, children and older adults may be affected.
Viral, fungal, and parasitic infections as well as bacterial infections may be found.
The serum concentration of IgM is normal in about half of the patients.
Abnormalities in T cell numbers or function are common. The majority of patients have normal numbers of B cells; however, some have low or absent B cells.
Approximately 50% of patients have autoimmune manifestations. There is an increased risk of malignancy.
Differential diagnosis of hypogammaglobulinemia includes drug-induced, for example secondary to glucocorticoids (steroids).
Diagnostic Criteria for Primary Immunodeficiencies. Mary Ellen Conley, Luigi D. Notarangelo, and Amos Etzioni Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clinical Immunology, Vol. 93, No. 3, December, pp. 190–197, 1999.
Common Variable Immunodeficiency. eMedicine.
Common Variable Immunodeficiency: A Multifaceted And Puzzling Disorder. Expert Review of Clinical Immunology, Medscape, 2009.
Reviewer: S. Randhawa, M.D., Allergist/Immunologist and Assistant Professor at NSU
A 54-year-old Caucasian male (CM) is evaluated by the Allergy and Immunology clinic for recurrent sinusitis for the last 20-30 years with 5-6 episodes per year. His IgG level was 470 mg/dL two months ago and IgM and IgA levels were normal. He reports no other significant infections. The patient reports seasonal exacerbations of his nasal symptoms in the spring, fall and winter. His last sinus infection was 9 months ago and he has not had such a recurrence free period in years.
Past medical history
Hypertension (HTN), hyperlipidemia (HLP), sinusitis, non-allergic rhinitis (non-AR), dermatographism.
Medications
Fexofenadine, flunisolide nasal spray, lisinopril.
Physical examination
Normal.
What is the most likely diagnosis?
Common Variable Immunodeficiency (CVID).
Allergic or non-allergic rhinitis.
What tests would you order?
Pre- and post-pneumococcal immunization titers.
ANA, HIV, RF.
B and T-cell flow cytometry.
RAST to aeroallergens.
What happened?
Skin prick testing: uninterpretable due to dermatographism. RAST to aeroallergens: negative.
HIV, ANA, RF were negative. CXR and U/A without evidence of malignancy. He had a colonoscopy in the past 3 years and it was negative.


Pre-immunization titers (left). Post-immunization titers (right) (click to enlarge the images).
Pre- and post-pneumococcal immunization titers 4 weeks apart showed an insufficient increase in the antibody titers: only 30% (4 out of 13 titers) of specific antibodies increased by the magnitude of 2 fold.
Flow Cytometry. Specimen: Peripheral Blood. Processing: Lyse, cytospin, stain with monoclonal antibodies. Cell Count: 8.2 10*6 total cells. Viability: 89%.
Total events analyzed = 42415
A gate Bright CD45 positive events = 5758 (17% of total events analyzed)
| CD45+ | 100% |
| CD19+/CD20 | 3% |
| CD19+/CD38 | 2% |
| CD19+/CD10 | less than 2% |
| CD3+/CD5+ | 85% |
| CD3+/CD7+ | 82% |
| CD3+/CD4+ | 60% |
| CD3+/CD8+ | 22% |
| CD3-/CD56+ | 11% |
| Total CD25 | 9% |
| CD25+/CD3+ | 9% |
| CD25+/CD5+ | 9% |
Interpretation: Lymphocytes comprise 17% of the total events analyzed, with 3% B-cells, 85% T-cells, and 11% NK cells. The CD4:CD8 ratio is 2.7:1. 9% of the lymphocytes are CD25 (dim) positive T-cells.
In healthy subjects, B cells constitute 18-47% of the peripheral lymphocytes. B cell count can be either low or normal in patients with CVID.
What happened next?
The patient was diagnosed with common variable immunodeficiency (CVID).
Would you start intravenous immunoglobulin G (IVIG) therapy for this patient?
Historically, the level of IgG of 470 mg/dL did not typically require replacement with IVIG in the absence of severe recurrent infections. Some of the newer studies point to an optimal replacement level of IgG of 600-800 mg/dL.
We decided to repeat the quantitative serum immunoglobulins in 4-6 months and to decide about IVIG replacement at that point.
Final diagnosis
Common Variable Immunodeficiency (CVID).
What did we learn from this case?
Evaluation of the response to pneumococcal vaccination is best accomplished by comparing pre-vaccination and post-vaccination antibody levels. A 2 to 4 fold increase in type-specific antibodies measured 4-6 weeks after vaccination is expected in immunocompetent adults. The number of serotypes for which a 2 to 4 fold increase is observed varies greatly among individuals; a consensus panel has suggested that individuals older than 5 years should respond to at least approximately 70% of pneumococcal serotypes. Adults older than 65 years may exhibit a smaller (less than 2 fold) increase in type specific antibody levels.
The "rule of thumb" is that 2/3 of titers have to increase 2-3 fold.
The patient did not have evidence of allergen sensitization on ImmunoCAP test for aeroallergens, therefore the intranasal steroids and oral antihistamine were discontinued. He was advised to use nasal saline rinses BID.
What are the normal serum immunoglobulin levels (IgG, IgA, IgM)?
Serum levels of IgM, IgG and IgA vary with age, gender and race.
The IgG and IgA concentrations in children show a gradual rise with increasing age. The IgA level is generally about the same in both sexes. Girls typically have higher IgM and IgG levels than boys.
The confidence interval bounded by two standard deviations about the mean excludes 5% of apparently healthy controls.
Elevated IgM, low IgA, low IgG, low IgM, and elevated IgA are the commonest changes observed in apparently healthy humans.
Humoral immunodeficiency is commonly defined as IgG, IgM or IgA level that is two standard deviations (2 SD) below the mean level for IgG, IgM or IgA, respectively, for the particular age group and gender.
Serum levels of IgM, IgG and IgA. Source: Pediatrics, 1966 and Immunologic disorders in infants and children, by E. Richard Stiehm, Hans D. Ochs, Jerry A. Winkelstein.
Typical pattern of immunoglobulin levels (IgG, IgA, IgM) in humoral immunodeficiency. Click here to enlarge the table.
What is a normal response to pneumococcal immunization?
Protection against infection and colonization is associated with specific IgG concentrations of 1.3 mcg/ml.
A second component in judging response is the final concentration of antibodies after immunization regardless of increase from preimmunization concentration. An adequate response is a post-immunization antibody concentration of more than 1.3 mcg/ml, even in the absence of a fourfold increase.
Children 2 to 5 years of age are normally expected to have an adequate response to more than 50% of serotypes tested. Patients 6 years or older are normally expected to respond to more than 70% of the serotypes tested. Source: Assessment and clinical interpretation of polysaccharide antibody responses. Kenneth Paris and Ricardo Sorensen. Annals of Allergy, Asthma, and Immunology, 2007; volume 99, Issue 5, Pages 462-464, and AAAAI Ask the Expert, 2012.
References
Serum immunoglobulin levels in healthy children and adults. J. W. Stoop, B. J. M. Zegers, P. C. Sander, and R. E. Ballieux. Clin Exp Immunol. 1969 January; 4(1): 101–112.
The relationship of race, sex, and age to concentrations of serum immunoglobulins expressed in international units in healthy adults in the USA. S. E. Maddison, C. C. Stewart, C. E. Farshy, and C. B. Reimer. Bull World Health Organ. 1975; 52(2): 179–185.
Serum immunoglobulin concentrations in preschool children measured by laser nephelometry: reference ranges for IgG, IgA, IgM. D Isaacs, D G Altman, C E Tidmarsh, H B Valman, and A D Webster. J Clin Pathol. 1983 October; 36(10): 1193–1196.
D Isaacs, A D Webster, and H B Valman. Clin Exp Immunol. 1984 November; 58(2): 335–340.
Serum Immunoglobulin Levels Throughout the Life-Span of Healthy Man. Ann of Int Med, November 1, 1971, Vol. 75 no. 5 673-682.
Diagnostic Criteria for Common Variable Immunodeficiency (CVID): Probable and Possible Diagnosis
The diagnostic criteria are divided into three categories: definitive, probable, and possible. There are no criteria for definitive diagnosis of Common Variable Immunodeficiency (CVID) at this time.
To guard against the inclusion of patients who have polymorphic variants in the genes associated with immunodeficiency and to specify the clinical or laboratory finding that is most consistently abnormal in a particular disorder, the patient must fulfill an inclusion criterion that is characteristic of the disorder.
Definitive diagnosis
Patients with a definitive diagnosis are assumed to have a greater than 98% probability that in 20 years they will still be given the same diagnosis. Mutation detection is the most reliable method of making a diagnosis but a single mutation is rarely found in CVID.
Probable diagnosis
Patients with a probable diagnosis are those with all of the clinical and laboratory characteristics of a particular disorder but who do not have a documented abnormality in the gene, the mRNA, or the protein that is known to be abnormal in the disorder. They are assumed to have a greater than 85% probability that in 20 years they will be given the same diagnosis.
Probable diagnosis of CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in serum IgG AND IgA and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Possible diagnosis
Patients with a possible diagnosis are those that have some but not all of the characteristic clinical or laboratory findings of a particular disorder.
Possible diagnosis if CVID:
Male or female patient who has a marked decrease (at least 2 SD below the mean for age) in one of the major isotypes (IgM, IgG, and IgA) and fulfills all of the following criteria:
1. Onset of immunodeficiency at greater than 2 years of age.
2. Absent isohemagglutinins and/or poor response to vaccines.
3. Defined causes of hypogammaglobulinemia have been excluded
Clinical features of CVID
Most patients with CVID are diagnosed with immunodeficiency in the second, third, or fourth decade of life, after they have had several pneumonias; however, children and older adults may be affected.
Viral, fungal, and parasitic infections as well as bacterial infections may be found.
The serum concentration of IgM is normal in about half of the patients.
Abnormalities in T cell numbers or function are common. The majority of patients have normal numbers of B cells; however, some have low or absent B cells.
Approximately 50% of patients have autoimmune manifestations. There is an increased risk of malignancy.
Differential diagnosis of hypogammaglobulinemia includes drug-induced, for example secondary to glucocorticoids (steroids).
References
Diagnostic Criteria for Primary Immunodeficiencies. Mary Ellen Conley, Luigi D. Notarangelo, and Amos Etzioni Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clinical Immunology, Vol. 93, No. 3, December, pp. 190–197, 1999.
Recognizing Primary Immune Deficiency in Clinical Practice. Clinical and Vaccine Immunology, March 2006, p. 329-332, Vol. 13, No. 3.
Common Variable Immunodeficiency. eMedicine.Common Variable Immunodeficiency. eMedicine.
Common Variable Immunodeficiency: A Multifaceted And Puzzling Disorder. Expert Review of Clinical Immunology, Medscape, 2009.
Interpretation of pneumococcal antibody titers. AAAAI, Ask the Expert, 2011.
Assessment of antibody response to pneumococcus immunization. AAAAI, Ask the Expert, 2011.
Heptavalent pneumococcal conjugate vaccine (PCV7) targets only 7 of the more than 92 pneumococcal serotypes. Lancet, 2011.
Cost-effectiveness of Pneumococcal Conjugate Vaccine vs. Polysaccharide Vaccine in Adults: PCV13 was better than PPSV23. JAMA, 2012.
Published: 02/06/2009
Updated: 02/07/2012
Heptavalent pneumococcal conjugate vaccine (PCV7) targets only 7 of the more than 92 pneumococcal serotypes. Lancet, 2011.
Cost-effectiveness of Pneumococcal Conjugate Vaccine vs. Polysaccharide Vaccine in Adults: PCV13 was better than PPSV23. JAMA, 2012.
Published: 02/06/2009
Updated: 02/07/2012
Subscribe to:
Posts (Atom)